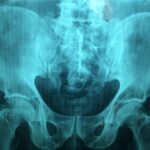
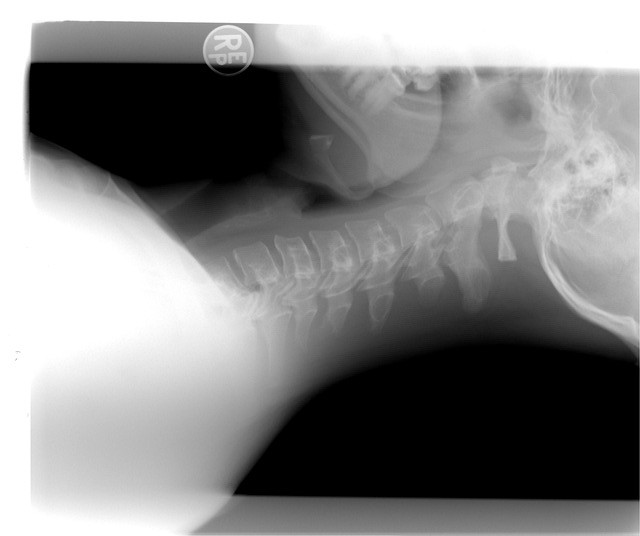
Albers-Schönberg disease, also known as adult autosomal dominant osteopetrosis or marble bone disease, is a hereditary disorder characterised by increased bone density throughout the skeleton. Although the bones appear markedly dense on imaging, their structure is abnormal, and they may be brittle, leading to fractures, bone pain, and complications related to the excessive bone mass. Early recognition and management of complications are important to preserve function and quality of life.
Pathophysiology and Biology
The underlying problem in Albers-Schönberg disease lies in impaired bone resorption. Normally, bone is remodelled by a balance of bone formation and breakdown; in this condition, the cells responsible for bone breakdown (osteoclasts) are dysfunctional or decreased in number. As a result, old bone is not removed as new bone is formed, leading to the accumulation of dense, sclerotic bone.
Despite appearing stronger, the internal architecture of bone is disrupted, the medullary canals become narrowed or obliterated, the bone marrow space is reduced, and the micro-structure of trabecular bone is disordered. These changes increase the risk of fracture, deformity, and compromise of marrow or nerve function.
Epidemiology
This form of osteopetrosis is the more moderate, adult-onset variant and is inherited in an autosomal dominant manner. It is sometimes discovered incidentally when dense bones are seen on imaging. Although the exact prevalence is variable, it is considerably rarer than common bone disorders. Many affected individuals live into adulthood and may have near-normal life expectancy, especially when complications are minimal.
Clinical Features
Symptoms
Symptoms vary widely; some individuals remain largely asymptomatic and are diagnosed only after imaging for unrelated issues. Others experience symptoms including bone pain, recurrent fractures (often from low-energy trauma), back pain, hip and knee arthritis, and problems related to high bone density.
Physical Examination
On examination, skeletal findings may include increased bone density visible on imaging, possible skeletal deformities, hearing or vision changes if nerve foramina are narrowed, and signs of bone marrow compromise such as anaemia or extramedullary hematopoiesis in more advanced cases. Joint mobility may be affected, and bone deformities may cause altered mechanics.
Diagnosis
Diagnosis is typically based on clinical features and characteristic imaging. Plain radiographs often show diffuse osteosclerosis (increased bone density), “bone-within-bone” appearance (endobone), sandwich-vertebra appearance in the spine, and narrowing of medullary canals. Laboratory tests may show marrow suppression or associated blood count abnormalities in severe cases. Genetic testing may confirm underlying mutations in genes affecting osteoclast function.
Treatment
While there is no cure that reverses the bone changes completely in all cases, management is tailored to symptoms and complications, with goals of reducing fracture risk, preserving function, treating complications, and monitoring for associated issues.
Non-surgical management emphasises bone health optimisation, fracture prevention, joint protection, physiotherapy, and monitoring for complications such as marrow failure or nerve compression. Surgical interventions may be required for fractures, joint arthrosis, or nerve decompression when indicated. Haematologic issues may demand specialist care. In severe early-onset forms of osteopetrosis, bone marrow transplantation may be considered, but its role in adult-onset Albers-Schönberg disease is limited.
Prognosis and Complications
Prognosis in the adult form is generally better than in the infantile variants; many individuals maintain good function and a normal lifespan. However, complications can occur, including pathological fractures, osteoarthritis of major joints, anaemia and extramedullary haematopoiesis, cranial nerve compression (leading to hearing or vision loss), bone infections such as osteomyelitis, and impaired mobility from deformities or recurrent injury. Lifelong monitoring is advisable to detect and address these issues early.
Prevention and Patient Education
Because the condition is inherited, genetic counselling may be appropriate for affected individuals or families. Patients should be educated about fracture risk, joint protection, avoiding excessive loads, maintaining bone health (nutrition, physical activity suited to condition), and reporting symptoms such as bone pain, changes in hearing or vision, or signs of marrow suppression. Early therapy of fractures or complications and regular monitoring of bone, joint, and hematologic health help preserve quality of life.
References
Root, JH. (1935). Albers-Schönberg’s Disease. American Journal of Diseases of Children, 49(4), 964-973.
Whyte, MP. (2023). Osteopetrosis: discovery and early history of “marble bone disease”. Bone, 171, 116737.
Pyeritz, R. E. (2025). Disorders of Bone Density, Volume, and Mineralization. In Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics (pp. 91-116). Academic Press.