Introduction
Beals syndrome, also known as Congenital Contractural Arachnodactyly (CCA), is a rare genetic connective tissue disorder. It was first described by Dr. Rodney Beals in 1972 and is often compared to Marfan syndrome because of similarities in physical appearance, such as tall stature, long limbs, and joint abnormalities. However, Beals syndrome has its own distinctive features, most notably joint contractures (tightness of joints) and “crumpled” ear deformities (1).
The condition is caused by mutations in the FBN2 gene, which plays a crucial role in the development of connective tissue (1). Although Beals syndrome can present challenges in mobility and physical growth, many individuals with proper medical care and therapy can lead fulfilling lives.
Symptoms
The symptoms of Beals syndrome vary in severity from person to person. Some children show obvious signs at birth, while others may develop more visible features during early childhood (2). The hallmark features include:
- Joint Contractures: Stiffness and tightening in joints, particularly in the knees, elbows, hips, and ankles. These can limit mobility and affect walking or posture.
- Arachnodactyly (long, slender fingers and toes): Similar to Marfan syndrome, the fingers may appear unusually long and thin.
- Crumpled ears: A distinctive feature where the outer ear looks folded or wrinkled.
- Long limbs and tall stature: Many affected individuals have disproportionately long arms and legs compared to their torso.
- Scoliosis (curvature of the spine): Progressive spinal curvature that may worsen with age.
- Muscle underdevelopment: Some children experience reduced muscle mass, leading to a thinner appearance.
- Facial features: Narrow face, high-arched palate, and sometimes a small jaw.
In rare cases, individuals may also have heart or eye abnormalities, though these are less frequent compared to Marfan syndrome.
Causes
Beals syndrome is primarily caused by mutations in the FBN2 gene located on chromosome 5. The FBN2 gene provides instructions for producing fibrillin-2, a protein essential for the formation of elastic fibers in connective tissue. When the gene is mutated, fibrillin-2 is either defective or insufficient, leading to abnormal connective tissue development.
This defective connective tissue is what causes the characteristic signs, including joint contractures, long limbs, and ear deformities.
Risk Factors
Beals syndrome is an autosomal dominant disorder, meaning only one copy of the altered gene is enough to cause the condition (2). The risk factors include
- Family history: If one parent has Beals syndrome, there is a 50% chance of passing it on to their child.
- Genetic mutations: In some cases, Beals syndrome arises from a new mutation in the FBN2 gene with no family history.
There are no environmental or lifestyle-related risk factors since it is strictly a genetic condition.
Diagnosis
Diagnosing Beals syndrome can sometimes be challenging because of its resemblance to Marfan syndrome (2). A proper diagnosis usually involves:
- Physical Examination: Doctors look for joint contractures, crumpled ears, scoliosis, and other distinct features.
- Family History: A genetic background check helps identify inheritance patterns.
- Genetic Testing: The most definitive method of diagnosis, testing for mutations in the FBN2 gene.
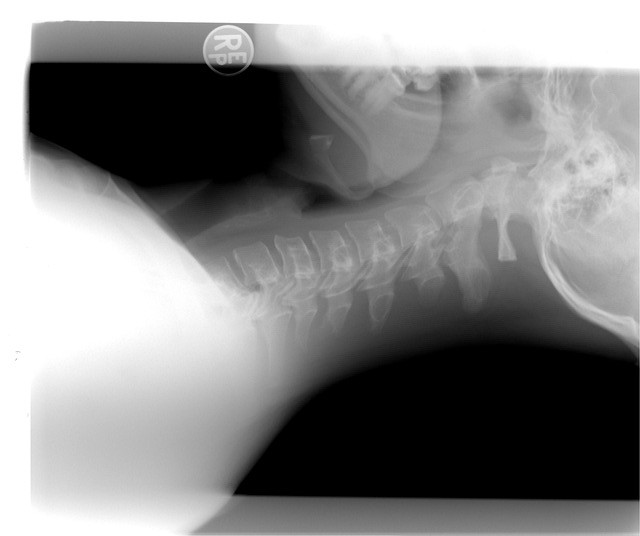
- Imaging Tests: X-rays or MRI may be recommended to assess skeletal abnormalities like scoliosis or joint issues.
- Cardiac Evaluation: Although less common than in Marfan syndrome, some patients may undergo echocardiography to rule out heart defects.
Treatment Options
Currently, there is no cure for Beals syndrome. Treatment focuses on managing symptoms, improving mobility, and preventing complications (3). The options include
- Physical Therapy: Regular stretching and strengthening exercises to improve joint flexibility and reduce contractures.
- Occupational Therapy: Helps children and adults adapt daily activities to maintain independence.
- Orthopedic Interventions:
- Braces or splints for joint support.
- Surgery may be considered in severe cases of scoliosis or contractures.
- Pain Management: Over-the-counter or prescribed medications to relieve discomfort from joint stiffness.
- Cardiac and Eye Monitoring: Though less common, periodic check-ups ensure early detection of any related complications.
- Genetic Counseling: For families planning to have children, counseling can help understand inheritance risks and options.
Living With or Prevention
Since Beals syndrome is a genetic condition, there is currently no way to prevent it. However, individuals and families can take proactive steps to manage the condition and improve quality of life.
Living with Beals syndrome involves
- Early intervention: Beginning physical therapy at a young age can significantly improve mobility and reduce long-term joint stiffness.
- Regular check-ups: Orthopedic, cardiac, and ophthalmologic evaluations help prevent or manage complications.
- Adaptive lifestyle: Some individuals may need modifications in school or work environments to accommodate mobility challenges.
- Emotional and social support: Counseling or support groups can help children and families cope with the psychological impact of living with a rare genetic condition.
For families with a history of Beals syndrome, genetic testing during pregnancy or before conception (through IVF with genetic screening) may provide insights into risk and help in family planning (3).
References
- Viljoen D. Congenital contractural arachnodactyly (Beals syndrome). J Med Genet. 1994 Aug;31(8):640-3. doi: 10.1136/jmg.31.8.640. PMID: 7815423; PMCID: PMC1050028.
- Hu L, Li H, Sun G, Wu K, Luan Z, Xiang Y, Tang S. Mutation analysis and prenatal diagnosis of a family with congenital contractural arachnodactyly. Mol Genet Genomic Med. 2021 Apr;9(4):e1638. doi: 10.1002/mgg3.1638. Epub 2021 Feb 27. PMID: 33638605; PMCID: PMC8123754.
- Chen L, Diao Z, Xu Z, Zhou J, Wang W, Li J, Yan G, Sun H. The clinical application of preimplantation genetic diagnosis for the patient affected by congenital contractural arachnodactyly and spinal and bulbar muscular atrophy. J Assist Reprod Genet. 2016 Nov;33(11):1459-1466. doi: 10.1007/s10815-016-0760-y. Epub 2016 Jul 9. PMID: 27393415; PMCID: PMC5125142.